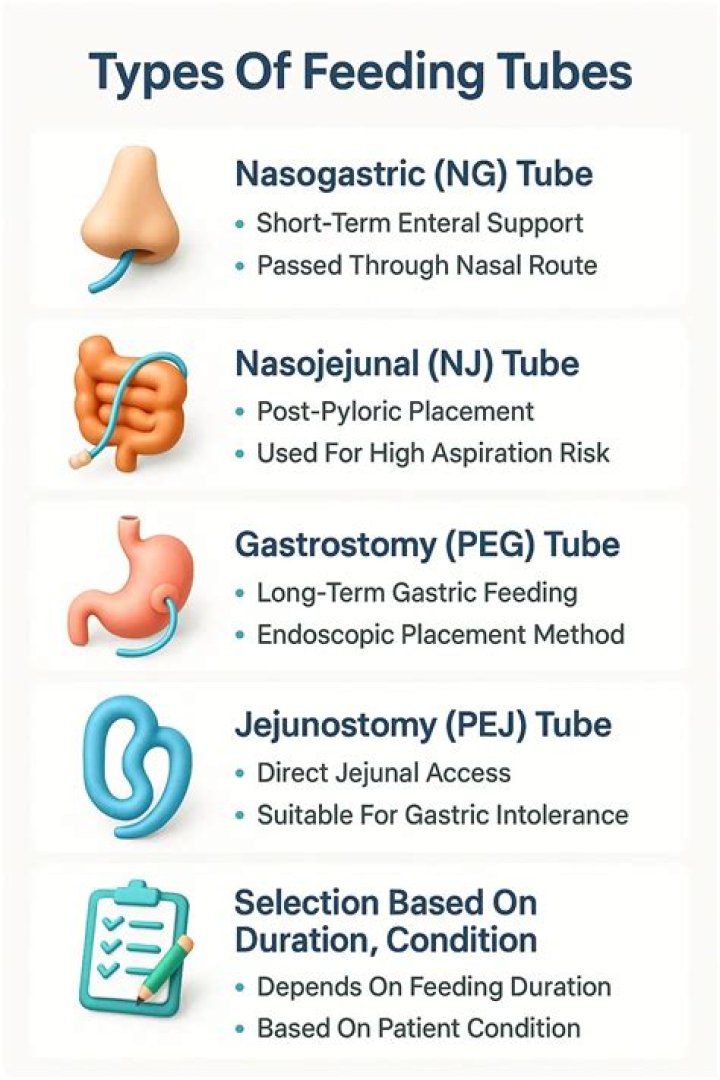
Sequence alignments are useful in bioinformatics for identifying sequence similarity, producing phylogenetic trees, and developing homology models of protein structures. However, the biological relevance of sequence alignments is not always clear.
Why sequence alignment is important for evolutionary study?
An ever-increasing number of evolutionary studies depend on the assembly of accurate multiple sequence alignments. Most alignment-based methods of evolutionary inference make an implicit assumption that the data represents the real evolutionary relationships between characters i.e. that the alignment is correct.
Why multiple sequence alignment is important?
Multiple sequence alignment (MSA) has assumed a key role in comparative structure and function analysis of biological sequences. It often leads to fundamental biological insight into sequence-structure-function relationships of nucleotide or protein sequence families.
What is the purpose of alignment in bioinformatics?
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences.Which alignment is useful to detect the highly similar sequences?
Conclusion: Quasi-alignment-based algorithms can detect highly similar regions and conserved areas across multiple sequences.
Why is protein sequence alignment produce more reliable result than DNA sequence alignment?
The simple fact that proteins are built from 20 amino acids while DNA only contains four different bases, means that the ‘signal-to-noise ratio’ in protein sequence alignments is much better than in alignments of DNA.
What is sequence alignment algorithm?
The alignment algorithm is based on finding the elements of a matrix where the element is the optimal score for aligning the sequence ( , ,…, ) with ( , ,….., ). Two similar amino acids (e.g. arginine and lysine) receive a high score, two dissimilar amino acids (e.g. arginine and glycine) receive a low score.
Is muscle better than Clustalw?
Published tests show that MUSCLE can achieve both better average accuracy and better speed than CLUSTALW or T-Coffee, depending on the chosen options.How can a DNA or protein alignment be used in species analysis?
DNA or protein alignment is a computational technique used to line up similar sequences between DNA or proteins in two different but potentially related species. This allows for the identification of differences between the two sequences. … This can be useful in an evolutionary species analysis.
What is the motivation behind multiple sequence alignments?Thus, instead of aligning two sequences, the objective in MSA is to align k sequences simultaneously such an overall functional is optimized. The motivation behind doing a MSA is that it allows us to extract consensus evident in a widely diverse set of sequences.
Article first time published onWhich alignment is useful to detect the highly conserved region?
Conclusion. Quasi-alignment-based algorithms can detect highly similar regions and conserved areas across multiple sequences.
How do you do sequence alignment?
- Click on the Align link in the header bar to align two or more protein sequences with the Clustal Omega program.
- Enter either protein sequences in FASTA format or UniProt identifiers into the form field (Figure 39)
- Click the ‘Run Align’ button.
What is sequence identity?
Sequence identity is the amount of characters which match exactly between two different sequences. Hereby, gaps are not counted and the measurement is relational to the shorter of the two sequences.
What is sequence alignment problem?
The Sequence Alignment problem is one of the fundamental problems of Biological Sciences, aimed at finding the similarity of two amino-acid sequences. Comparing amino-acids is of prime importance to humans, since it gives vital information on evolution and development.
What are the advantages of using a protein sequence rather than a DNA sequence?
In contrast, protein sequence is composed of 20 characters (AA). The sensitivity of the comparison is improved. It is accepted that convergence of proteins is rare, meaning that high similarity between two proteins always means homology. The DNA databases are much larger, and grow faster than protein databases.
Why might it be a better idea to align by amino acid sequence rather than DNA?
Second, because amino acids are more conserved evolutionarily than DNA, and possibly because the amino-acid alphabet is larger than the DNA one and therefore less likely to become saturated with convergent substitutions over longer timeframes, it is often easier to align amino-acid sequences between more distantly …
Why is amino acid sequence important?
The linear sequence of amino acids within a protein is considered the primary structure of the protein. … The chemistry of amino acid side chains is critical to protein structure because these side chains can bond with one another to hold a length of protein in a certain shape or conformation.
What is biological sequence alignment?
Sequence alignment is a way of arranging protein (or DNA) sequences to identify regions of similarity that may be a consequence of evolutionary relationships between the sequences. From: Encyclopedia of Bioinformatics and Computational Biology, 2019.
How does MUSCLE sequence alignment work?
MUSCLE uses the sum-of-pairs (SP) score, defined to be the sum over pairs of sequences of their alignment scores. The alignment score of a pair of sequences is computed as the sum of substitution matrix scores for each aligned pair of residues, plus gap penalties.
What is the difference between MUSCLE and clustal alignment?
ClustalW implements an iterative algorithm so mistakes produce in earlier step are quite unlikely to be corrected in later step meanwhile muscle implements an progressive algorithm allowing re-optimizations of columns during the whole process.
How many sequences can MUSCLE align?
MUSCLE stands for MUltiple Sequence Comparison by Log- Expectation. MUSCLE is claimed to achieve both better average accuracy and better speed than ClustalW2 or T-Coffee, depending on the chosen options. Important note: This tool can align up to 500 sequences or a maximum file size of 1 MB.
How can identifying conserved sequences help researchers?
Conserved sequences help us find homology (similarity) among different organisms and species. Phylogenetic relationships and trees could be developed and effective ancestry could be found using the data on conserved sequences.
Why are highly conserved regions important?
Why are highly conserved regions important? Highly conserved regions are some parts of a gene that are extremely similar among different species. They are important because universal primers bind to them so that they can be used to copy DNA from a variety species of bacteria.
How do you know if a sequence is conserved?
Conserved sequences may be identified by homology search, using tools such as BLAST, HMMER, OrthologR, and Infernal. Homology search tools may take an individual nucleic acid or protein sequence as input, or use statistical models generated from multiple sequence alignments of known related sequences.
How long does sequence alignment take?
For instance, the sequencing program MUSCLE can usually handle large data sets with a premium on accuracy. For some perspective, I can usually align ~750 sequences of 1000 nucleotides each in about an hour using MUSCLE. For aligning a large number of sequences, you must have sufficient computer memory and storage.
What is sequence similarity in bioinformatics?
Sequence similarity is a measure of an empirical relationship between sequences. … A similarity score is therefore aimed to approximate the evolutionary distance between a pair of nucleotide or protein sequences.
Why is it useful to search a database to identify sequences that are homologous to a newly determined sequence?
Discuss why it is useful to search a database to identify sequences that are homologous to a newly determined sequence. By searching a database, you can identify genetic sequences that are homologous to a newly determined sequence. In most cases, homologous sequences carry out identical or very similar functions.
What is sequence identity in bioinformatics?
Sequence identity refers to the occurrence of exactly the same nucleotide or amino acid in the same position in aligned sequences.